English / Japanese
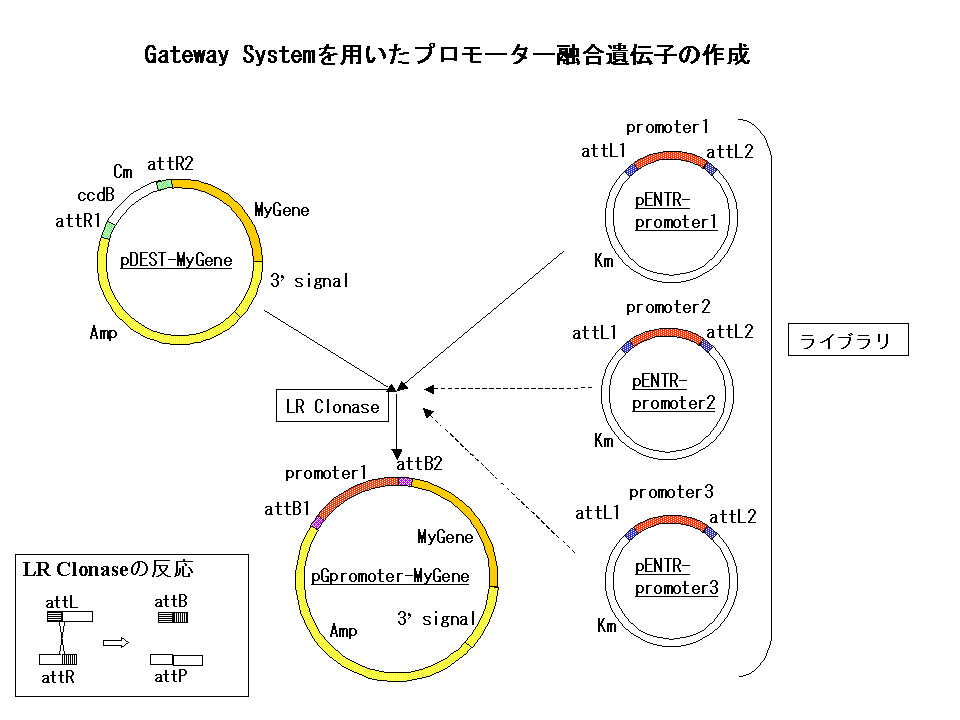
pENTR-promoter プラスミドの作成原理図 pDEST-MyGene プラスミドの作成原理図
特定の組織、細胞に手持ちの遺伝子を発現させるためのプロモーター付加遺伝子の作成を簡便化するために、Gatewayシステムを採用しています。以下、その概略を説明します。
1)特定の細胞で発現を誘導することのわかっているプロモーターをpENTRベクターにクローニングします。発現パターンが予定した通りであることを確認します(*)。pENTRベクターにクローニングしたプロモーターのコレクションは汎用ライブラリーとして、さまざまな遺伝子の発現のために用います。
2)発現させたい遺伝子(MyGeneと呼ぶ)はpDESTベクターにクローニングします。
3)pENTRプラスミドとpDESTプラスミドとに、組換え酵素であるLR Clonaseを作用させることにより、部位特異的組換えが起こり、MyGeneにプロモーターがつながった形のプラスミドが正確に作り出されます。
4)このプラスミドを線虫に導入して発現実験を行います。
(*)あらかじめpDEST-GFP, pDEST-CYP, pDEST-YFP, pDEST-Venus, pDEST-DsRedなどが作成されていますので、作成したpENTR-プロモータープラスミドは、最初にpDEST-GFP等と組換えを行い、線虫に導入して正しい細胞で発現していることを確認します。
この方法の利点は、制限酵素を用いずに組換えを行うので、汎用性がある点です。これに対し、制限酵素を用いてプロモーターと遺伝子を結合する場合には、その制限酵素が他の部位を切断しないという条件があるので、いろいろなプロモーターを試す場合にはプロモーターごとに操作が異なり煩雑です。
また、プロモーターはゲノムからのPCRによりクローニングされるケースが多いですが、PCRによる変異等を考えると、一旦発現パターンを確認した上で、PCRを用いない方法によりサブクローニングすることが好ましいと考えられます。発現パターンの確認されたプロモーターをライブラリーとし、Gatewayシステムを用いてサブクローニングを行うことは、こういった意味でも有利です。
(注)本システムにおける「プロモーター」は翻訳開始コドンATGを含みません。一方、MyGeneはATGを含む形でデザインする必要があります。この方式は、「ORFeome
project」(Reboul et al., Nature Genetics 34: 35-41 2003 )と整合性を欠くという欠点があります。しかし、本システムは線虫の特定細胞での発現により遺伝子機能を解析するような実験を念頭において作られていますので、attB配列由来の8アミノ酸が遺伝子産物に付加されることを避けるため、敢えてATGを含まないプロモーターを用いています。
現在までに作成されているプラスミドについてはこちらのデータベースをご覧下さい。
Gateway Systemの原理 (詳しくはインビトロジェン社ホームページ参照)
λファージが大腸菌の染色体に挿入され、あるいは切り出されるときに、attB,
attPなど特定の塩基配列で組換えが起こることを利用しています。組換え酵素"BP
Clonase"(インビトロジェン社より販売、cat. 11789-013)はattB1+attP1→attL1+attR1およびattB2+attP2→attL2+attR2の組換え反応を触媒し、組換え酵素"LR
Clonase"(インビトロジェン社より販売、cat. 11791-019)はattL1+attR1→attB1+attP1およびattL2+attR2→attB2+attP2
の反応を触媒します。
プラスミド選択のための薬剤耐性マーカーとしてはAmp(アンピシリン)のほか、Cm(クロラムフェニコール)、Km(カナマイシン)が使い分けられています。また、逆選択マーカーとしてccdBが使われています。この遺伝子は通常の大腸菌には致死的に働くため、ccdBを持ったプラスミドはコロニーとして回収されません。ccdBをもつプラスミドを増やす際にはccdBに耐性なgyrA株(DB3.1、インビトロジェン
cat. 11782-018)を用います。